Newsletter | Spring 2016 ⋅ Number 69

Dr. Krystle J. McLaughlin is a Professor of Practice in the Department of Biological Sciences at Lehigh University.
Dr. McLaughlin received her BA in Physics from Colgate University in 2006. She went on to the University of Rochester where she received her Ph.D. in Biophysics, studying the structural and thermodynamic basis for protein-nucleic acid interactions using X-ray crystallography as he primary tool. Prior to Lehigh, she was a scholar in the SPIRE (Seeding Postdoctoral Innovators in Research and Education) program at University of North Carolina at Chapel Hill. As part of the SPIRE program, she worked in the Redinbo Lab where she continued to cultivate her love of structural research studying the spread of antibiotic resistance in Salmonella typhimurium, and also received pedagogy training, including in the effective use of active learning techniques into the classroom.
Dr. McLaughlin is very interested in engaging undergraduates through active and inclusive teaching methods in STEM education and outreach. She has created and facilitated several workshops for both undergraduates (e.g., How to prepare for research experiences) and graduate students (e.g., Strategies for Active Learning in the Classroom). She has also participated as an organizer and facilitator for faculty workshops at Lehigh (e.g. Inclusive Teaching). Additionally, Dr. McLaughlin is the director of the Pennsylvania DNA Day outreach program, which brings young scientists into high school classrooms to talk about DNA and genetics.
She can be contacted at krm514@lehigh.edu

Figure 1. Undergraduates examining protein secondary structure models (alpha-helix and beta-sheet) during biochemistry lab.
Lysozyme is my best friend in my ongoing mission to excite and teach students about protein structure. Within my undergraduate biochemistry lab, I introduced a module with several lab periods dedicated to exploring macromolecular X-ray crystallography: from protein purification techniques to building structures on the computer. Crystal growth is one of the more exciting parts of macromolecular crystallography, and here lysozyme shines, but I also use lysozyme to teach students more about molecular modeling and crystallization strategies. To help students have a better appreciation for protein structure and to understand the unique role of lysozyme in crystallography I use a combination of crystallization experiments, physical models and the PDB.
After an introduction to macromolecular crystallography in a short lecture, students crystallize lysozyme, using protocols from the literature.1 Lysozyme crystals grow very quickly, and when students see their crystals under the microscope for the first time there is a lot of excitement. We discuss how X-ray data is collected, and then the students are given preassembled physical models of protein secondary structure* to explore as a precursor to the next steps of investigating computational molecular models. Though during biochemistry lecture students are given images and characteristics of protein structure, the three-dimensional nature is not as easily grasped. When examining the alpha helix and beta sheet physical models (Figure 1) students worked in pairs to answer questions such as:
Where is the N-terminus? How do you know?
Can you figure out a repeating pattern that makes the α-helix?
Is this parallel or antiparallel beta-sheet? How do you know?
They struggled initially, but by the end were rewarded when they were able apply their theoretical knowledge. On end of semester feedback forms, one student wrote “It really helped me understand the structures of the molecules and it was something new that I didn't get to see in other labs.” The tactile nature of this part of the module facilitated a progression of learning for many of the students.
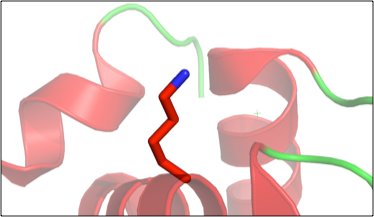
Figure 2. Example of student generated figure, using PyMol, showing the “fixed” amino acid in lysozyme (PDB 1VAU).
Next, the students head to the computer where they are given a modified lysozyme PDB entry file. Several residues in the lysozyme structure were mutated and the students must use the electron density map, which they obtain from the Electron Density Server using the PDB ID, to find the mutated residues, then infer from the map what the correct residue should be. Working in COOT,2 they “fix” the structure. Students really like this part as well, and during this exercise they are asked to see if they can recognize any secondary structure elements as they explore the lysozyme structure. This ties together the physical models with the computational model. When they are finished with this portion, they take their “fixed” (i.e., now wild type) lysozyme PDB file into PyMol,3 creating figures showing each modified amino acid (Figure 2). The module then concludes with a visit to the RCSB Protein Data Bank.4
On the RCSB PDB site, they navigate to the human dopamine receptor (PDB ID 3PBL), a transmembrane protein whose structure solution was aided by lysozyme. I tell the students that using protein engineering, scientists fused lysozyme to the dopamine receptor, purified the fusion protein, and crystallized the fusion protein- taking advantage lysozyme’s propensity to crystallize. They are instructed explore all the tabs on the PDB page for 3PBL to answer a series of questions including:
How many ligands are bound to the receptor? And what are their names?
What is the resolution of the crystal structure?
What method was used for crystallization?
What temperature was it crystallized at?
Was the data collected at a synchrotron?
What percentage of the structure is alpha helical? Made of beta sheets?
Students are often amazed by how many details they can learn about the structure just through exploring the PDB site. When they complete the questions they download the 3PBL PDB file, and open it in PyMol to answer one final question: Where is the lysozyme?
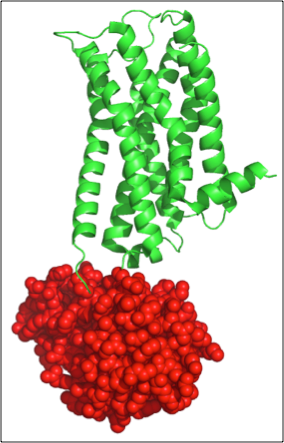
Figure 3. Example of student generated figure, using PyMol, to show lysozyme in red and the human dopamine receptor in green (PDB 3PBL).
Again, they are initially puzzled but eventually identify the two individual proteins, and display the lysozyme distinct from the dopamine receptor (Figure 3). When they find the lysozyme, there is always a lot of chatter and it stimulates thoughtful questions like: How is the lysozyme in the middle of the receptor and still folded individually? What follows are excellent discussions on protein domains, protein folding, and crystallization strategies.
A student who had moved on to a graduate biochemistry program reported back to me that skills taught in this lab, in particular understanding how to use the PDB and examine structures on the computer, turned out to be one of the most useful parts of her undergraduate education. There has been overwhelmingly positive feedback from the students both during and after this module. The combination of moving from the wet lab crystallization experiments, to physical structure models and finally to computational modeling seems to enhance student interest and appreciation of protein structure.
These words from a student on last year’s evaluations encapsulate my overarching goals: “I thought these labs showed us where the field was progressing. I had no idea that these things were possible on a computer.”