Newsletter | Fall 2016 ⋅ Number 71

Professor Todd Silverstein is a biochemist with over 30 years of full-time teaching experience in biochemistry, advanced biochemistry, and general chemistry. He has published 56 peer-reviewed papers, approximately two-thirds of which are pedagogical in nature; for several years he served as editor of the Journal of Chemical Education’s “Tested Demonstration” series. Dr. Silverstein’s research includes the study of biological membranes and bioenergetics (mitochondria and chloroplasts), and toxicological enzymology. He has an active undergraduate research group, having mentored seventy students since 1987.
Willamette University is a private liberal arts college located in Salem, Oregon, United States. Founded in 1842, it is the oldest university in the Western United States.
Training undergraduates in the use and manipulation of three-dimensional models of protein structure is not as rare as it used to be a decade ago,1-5 however, it is still not common. It is especially hard to find a project that examines these structures from the perspective of evolutionary relationships between homologous proteins.6 Accordingly, we developed a multi-week laboratory project in which students compare structures of myoglobin (Mb) and its homologues, and also isolate the protein and study its ligand binding and redox states. 7
The important laboratory techniques covered in this project include size-exclusion chromatography, electrophoresis, spectrophotometric titration, and FTIR spectroscopy. Regarding protein structure, students work with computer modeling (mainly PyMOL and Swiss Model) and FTIR to characterize both native and thermally denatured structures. Students also study protein function (ligand binding equilibrium), and are instructed on topics in data analysis (calibration curves, nonlinear vs. linear regression). This is a challenging and rewarding exercise that not only exposes students to a wide variety of important biochemical laboratory techniques, but also ties those techniques together to work with a single readily available and easily characterized protein, myoglobin.
Our extended laboratory project requires six 3-hour laboratory periods, which include some in-class time for data analysis. In laboratory period 1, students extract Mb from ground beef, reduce and oxidize portions of the sample, use a Sephadex column to “desalt” the excess oxidizing agent, and use UV-Vis absorbance to distinguish between the reduced and oxidized samples. In period 2, students employ SDS-PAGE to characterize the extracted samples. In period 3, azide binding to metMb is examined via spectrophotometric titration. In periods 4 and 5, students use online databases, websites, and software to characterize the structure of Mb and a selected novel homolog. In the final period, students employ FTIR spectroscopy to probe protein secondary structure in both the native and thermally denatured conformations.
The Protein Structure Laboratory Project
Here we describe in detail the myoglobin* structure modeling activities (periods 4 and 5). For a more complete description of the entire laboratory project, including laboratory manual instructions in the Supporting Information, see our Biochem. Molec. Biol. Educ. paper.7
In 1998, Leon et al. published a biochemistry laboratory project in which students used internet websites and freeware to model protein structure.6 Leon et al. described a project in which students selected a protein whose structure had already been solved and deposited in the Protein Data Bank (PDB). They then carried out a series of four operations on the internet: (a) using search programs (e.g., BLAST) to find a “novel homolog,” i.e., a homologous protein whose structure has NOT yet been solved; (b) submitting the amino acid sequence of the novel homolog to secondary structure prediction programs (e.g., JPRED4, PredictProtein) and comparing secondary structures of the original protein and its novel homolog ; (c) submitting the amino acid sequence of the novel homolog to Swiss Model to obtain a predicted three-dimensional structure; and finally (d) using PyMOL to compare the solved 3D structure of the original protein to the predicted structure of its novel homolog.
It takes students about 4.5 hours to finish operations (a – d) listed above. We have our students use the final 1.5 hours in laboratory period 5 to prepare figures for an oral presentation in which they carefully characterize the major differences and similarities between the original protein and its novel homolog.
All of the websites described by Leon et al. have changed dramatically in the nearly two decades since the original publication. Some things have gotten easier. For example, in 1998 one had to visit three separate websites to obtain a protein’s structure coordinates (PDB), structure and function information (UniProt/SwissProt), and perform homology searching (Blitz). Currently all of this information can be obtained from the RCSB PDB website and links therein. On the other hand, other aspects of the project have gotten more difficult. The protein visualization program recommended by Leon et al., Rasmac, is woefully outdated; in addition, a useful protein structure comparison program that we discovered around 2000, Protein Explorer, has not been supported since about 2006. However, an even better freeware program, PyMOL, has since come into wide use.5,8,9 This program not only allows homologous proteins to be aligned and visualized, but it also has many different viewing modes that can be implemented. We encourage students to explore the capabilities of PyMOL on their own, but we require that they use at least three different views to compare their two proteins: van der Waals surface; electrostatic surface; and polarity/hydrophobicity (Figure 3). The former two views are built into PyMOL, and the latter is provided by the “color_by_restype” script written by researchers at Queens University in Kingston, Ontario.10
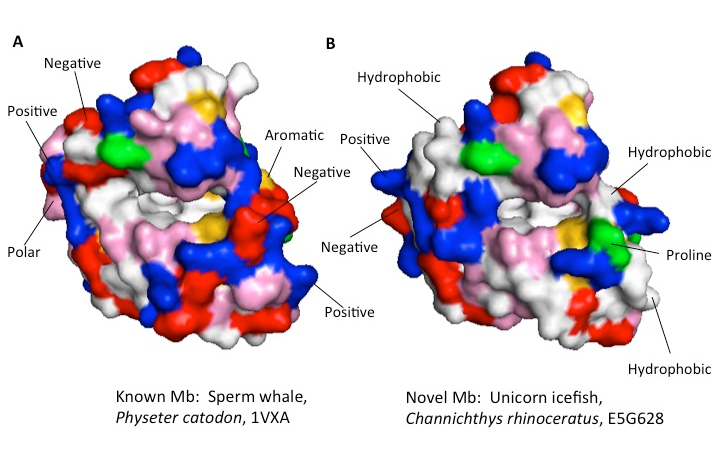
Figure 3: Mb structure comparison. A: solved structure of sperm whale Mb, PDB entry 1VXA; B: Swiss model predicted structure of Mb from unicorn icefish (Channichthys rhinoceratus), UniProt file E5G628. Color key for amino acid side chain types at the protein surface: white = hydrophobic; red = negative; blue= positive; pink = polar neutral; yellow = aromatic; green = proline. Note the heme binding cavity in the center of both structures; most of the surface directly above this cavity is identical in both A and B. Besides these and other similarities, a number of key differences between the two forms of Mb are marked above.
Student Report
For the protein structure section of the project, students prepare an oral presentation in which they carefully characterize the major differences and similarities between Mb and their chosen novel homolog. Students are expected to discuss not only gross differences in shape, polarity, and electrostatic charge, but also to identify the specific amino acids changes that are behind these differences. In the experimental part of the project, students use FTIR amide I peaks to distinguish between proteins that are primarily a-helix vs. b-sheet, and to follow temperature-driven changes in secondary structure. Finally, they use nonlinear regression of their FTIR peak vs. temperature data to determine ∆H° of unfolding and TU (T that yields 50% unfolding), and compare to literature values.
Further Extensions
We decided to focus our protein structure/function project on myoglobin because it is readily obtained (from ground beef, and also in purified form), easily characterized (by spectrophotometry), and has been used in a number of published biochemistry laboratory projects. There are undoubtedly many other proteins that fit this bill and could be substituted for Mb; we have also used, for example, lysozyme and lactate dehydrogenase.
Acknowledgments: We wish to thank Willamette University for its extensive long-term support of laboratory curriculum development; parts of this description are based upon work supported by the National Science Foundation under Grant No. DUE-1044737. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
* In order to increase the diversity of protein structures examined, we have half of the students start with myoglobin, a predominantly alpha-helical protein, and the other half start with the predominantly beta-sheet protein, lysozyme.