for Structural Bioinformatics Protein Data Bank
Spring 2009
Number 41
| Glossary About the Animators | Extended Interview PDB
COMMUNITY FOCUS Gaël McGill, Ph.D., and Graham Johnson, Molecular Animators Q. First off, can you tell me a bit about yourselves and the work you are doing in molecular graphics? A. GaëL McGill: Cell and molecular biology has been a passion from my middle school days. I came to the USA as an undergraduate specifically to study molecular biology and my dream was to be involved in research. I went on to do my Ph.D. at Harvard Medical School (mostly focusing on cancer signal transduction pathways and apoptosis using a varied mix of cell and molecular biology, biochemistry, animal model/genetics, and screening approaches). Having identified a need in the local academic, medical, biotech and pharmaceutical communities for "scientifically-informed" graphic design and web programming services, I started my company Digizyme (www.digizyme.com) in 1999 during my Ph.D. years. On a personal level, this really served as a creative outlet outside of the long hours at the bench. Digizyme has grown to offer more advanced services in recent years--including 3D animation services and even product design and visualization for the biomedical device industry. Over the past few years, I have "reintegrated" into academia in hopes of establishing a full-time team of scientist-animators at Harvard Medical School (where I currently teach Maya molecular visualization classes year-round). I enjoy the variety and challenge of client-driven projects as part of my work at Digizyme, and look forward to the freedom of pursuing larger-scale, longer-term collaborative projects relating to fundamental cell/molecular visualization challenges as an academic.
About the Animators A.Graham Johnson: I graduated from the Department of Art as Applied to Medicine in the Johns Hopkins School of Medicine with a masters degree in Medical and Biological Illustration in 1997. At Hopkins, we studied anatomy and physiology with medical school students while simultaneously sketching and illustrating our dissections along with autopsies and surgeries observed at the hospital. After graduating, I focused on studying molecular and cell biology while illustrating the textbook Cell Biology with Tom Pollard and Bill Earnshaw. I began animating this content for other clients and realized that trained medical illustrators could contribute a great deal to this relatively unillustrated subject. It occurred to me that one could simulate most of my animations either by scripting the physics or by translating imported data to a format that my 3D software package could recognize. I began simulating a handful of my client animations, but quickly learned that the out-of-the-box software was not intended for this purpose and could only be used for inaccurate and simple molecular interactions. In 2005, I applied to the Ph.D. program at The Scripps Research Institute to work in the Molecular Graphics Laboratory under Arthur Olson to better understand the content and to communicate directly with a team of talented molecular graphics coders. As I head into my fourth year of the program, Im finally producing some useable scripts that have made it easier to create illustrations and animations of molecular realms. I hope to distribute many of the tools very soon. Q. How do you import PDB structures? Are these tools generally available? A. Gaël McGill: For the most part, we use existing molecular graphics applications like Chimera and PyMOL to generate geometry files. These are typically exported as VRML (Virtual Reality Modeling Language) and then converted to OBJ format (a common data format for 3D data) before being imported into Maya. We also use MEL (Maya Embedded Language) scripts--either ones already available online (although currently there are not many related to PDB), or in-house/custom ones. Which method we use depends on what we will be doing with the geometry once inside Maya. A great option for bringing in large PDB datasets has been Chimera's "multiscale models" feature. Eventually it would be great to create a similar functionality for creating polygonal models within Maya itself in order to have more control over the output geometry. Still, this type of tool has been very useful in creating animations showcasing large complexes (like entire viruses). A. Graham Johnson: Ive written a COFFEE plug-in (Cinema4Ds native scripting language) that imports a single PDB file or a list of PDB files directly into my viewer window as a set of points in space (used to generate smooth surface models such as metaballs), CPK spheres, or backbone spline. Im building a primitive ribbon generator and hope to make the tools available for use within the next year. If I require a more sophisticated surface model, e.g., one colored by electrostatic potential, Ill export it from one of the popular molecular viewers as either a VRML or an OBJ file. Again, for static images, Ill often just export a screen grab from a molecular viewer that offers a style Im after.
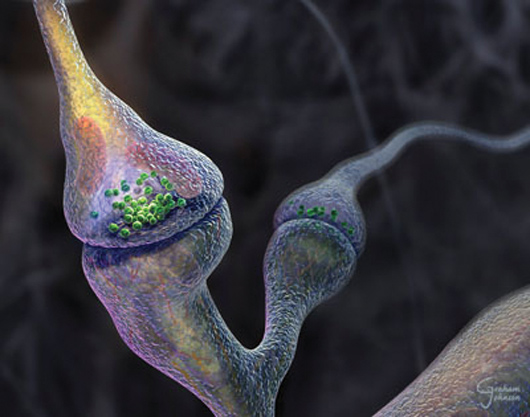
The Synapse Revealed created by Graham Johnson for the Q. What type of molecular imagery is most popular with your clients? A. Graham Johnson: Because molecular graphics viewers are so user-friendly these days, clients rarely come to me to request an image or movie of a single molecule spinning on a monochromatic background. Most of my clients ask me to generate an editorial image, or to illustrate or animate a process or cell event involving multiple molecule types. A. Gaël McGill: Although it depends on the project, I find that clients (especially biotech and pharma) want images of molecules "in context." In other words, scenery that captures a molecular process of interest but also places it within a cellular landscape. The challenge is to create a still image that captures or suggests a narrative or mechanism... essentially an "action shot" in which the visual context of the structure being depicted (and its binding partners) helps to communicate its function. Q. I imagine that assembly of biologically relevant complexes (such as chromatin or a transcription complex) and modeling their dynamics poses difficult challenges--what types of tools do you use for this? A. Gaël McGill: This is one of the toughest challenges at the moment (and it does not only apply just to complexes): how does one visually represent the dynamic aspects of proteins based on available (mostly static) data? The ability to create linear morphs between multiple conformational states of a protein using the adiabatic mapping technique (used by Mark Gerstein's method at www.molmovdb.org, for example) is very useful to visualize one possible trajectory, but it is only one possible trajectory and it also cannot tackle more complex morphs that involve partial refolding of protein domains. Drew Berry at The Walter and Eliza Hall Institute of Medical Research has pioneered a visual style that suggests the dynamics of proteins, but it would be nice to create animations that are based on actual data for these dynamics (i.e., as opposed to using noise/fractal motions throughout, having vibrations and degrees of flexibility that reflect the protein's actual range of 'thermodynamically-permissible' motion). In packages like Maya, we are currently limited to using pretty basic kinematic tools (i.e., building rigs driven by forward or inverse kinematics) that intrinsically have no knowledge of the molecular structure and its limitations or range of permissible torsion/bending. The software does not even register or warn against impending self-intersections--a problem that we are currently exploring in collaboration with topologists/software developers from the entertainment industry. At the moment (and depending on the target audience), we try to find as many sources of reference data as possible and use them as "inspiration" to create a dynamic representation of a protein or complex. The goal is to find more direct ways of integrating these data into the visualization (inasmuch as it helps communicate crucial parts of the story). A. Graham Johnson: Ive attempted to rig a handful of complex builders over the years with out-of-the-box toolsets. Such tools often do a great job of roughing a concept together, but fail when applied to large-scale systems or attempts to accurately simulate the rigor and detail often required for molecular imagery. Years ago, for example, I tried, to stitch together thousands of blocks with pairs of springs to represent the persistence length and flexible backbone of DNA in a plasmid. I animated a twist to see if it would supercoil, but the collision detector would always overload and the system would come to a screeching halt before the DNA could achieve a single twist. Ive tried pouring virtual molecules into virtual organelles to fill them with random recipes of non-colliding molecules, but again, the technique has always proved to be slow, limited in volume, and relatively uncontrollable. Most particle generators Ive toyed with have similar limitations to their physics simulation. To overcome these challenges, Ive begun to construct scripts from scratch that attempt to combine the capabilities of simplified molecular dynamics with the visualization power of commercial 3D software.
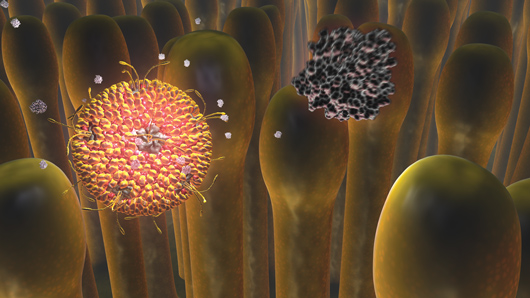
Early Events in Reovirus Entry by Gaël McGill. The full movie can be viewed online at www.molecularmovies.com/movies/mcgilliwasa_reovirus.html Q. Are there any resources that you would suggest to artists interested in incorporating PDB structures into their work? A. Gaël McGill: Other than the fantastic PDB itself (not sure what we would do without it!), I recently launched a free resource for scientists interested in learning 3D software packages for cell and molecular visualization at www.molecularmovies.org. One section is a showcase/directory of some of the web's best cell and molecular movies (organized by scientific topic), and another is dedicated to tutorials and lectures. There are currently hundreds of pages of free tutorials that approach learning Maya in the context of biological visualization. More specifically, several of these tutorials focus on getting PDB data into 3D applications like Maya. Expansion of the site in the near future will also include a "Toolkit" section where animators can share scripts and plugins for PDB import (and other tasks related to molecular animation), and a new section that provides a more general directory of visual resources. The idea behind this last section is to find and organize non "narrative-driven" raw data visualizations (i.e. like time-lapse movies, MD simulations and other datasets) that animators can use as reference materials to create better visualizations. A. Graham Johnson: The updated and integrated Electron Microscopy Data Bank (emdatabank.org) offers many low-resolution models of macromolecular structures and has a new online EM viewer. Many files in the PDB exist as low resolution structures with only alpha carbon coordinates published. If you need a rough approximation of the sidechains to generate a teaching model for such a molecule, you can generate or download a pre-generated version from MaxSprout (www.ebi.ac.uk/ Tools/maxsprout). Lastly, I find the TransMembrane PDB indispensable (pdbtm.enzim.hu). Q. Have you had any projects that posed an insurmountable challenge? A. Gaël McGill: The great thing about cell and molecular visualization is that there is an endless source of topics/mechanisms to visualize and each of these come with their own unique challenges. We may not always use the optimal solution or have the perfect tool available, but there is almost always a creative way to solve the visual representation challenges that emerge. It is one of the aspects of visualization with powerful packages like Maya that make this work so fresh and exciting! A. Graham Johnson: Many projects have and Ive often had to truncate my personal goals or compromise with the client to find some work-around because of strict deadlines. In years past, I sometimes had to resort to keyframe animation, hand drawn animation, simple 2D vector animation, and even static imagery to convey a message that could have been most clearly presented as a 3D animated sequence... I simply lacked the technology, skill, or time. Finding out, however, that molecular animation posed more challenges than my other medical illustration jobs directly inspired me to build tool sets to help meet such challenges. Q. What new tools would you like to have? A. Gaël McGill: As noted above, we are in the process of creating a suite of MEL scripts that can address some of the basic geometry-building tasks for getting PDB data into Maya (without having to resort to molecular graphics software-exported meshes). Once we have this first set of scripts (that just focus on efficient/clean geometry creation), the next step would be to explore the development of programmatically-driven rigging tools for defining the articulation of the models. In other words, to write scripts that not only create Maya-native geometry directly from the PDB but also automatically create a rig that has some inherent motion constraints applied. This is easier said than done and will of course depend on the type of molecular representation (ball & stick versus cartoon for example would have very different 'rules' applied to constrain motion). Having geometry that is more 'self-aware' (and that can at least avoid or warn about self-intersections) would be useful. A. Graham Johnson: I agree that methods for exporting molecular models in styles that are animation-ready would be very helpful to everyone in the molecular illustration field. I would primarily like to see an extension of the PDB that offers biological unit matrices to help users generate pertinent symmetries. This works great in PDB files for viruses that have BIOMT lines in REMARK 350 to describe the transformation and orientation matrices needed to generate a complete virus1. More specifically, Id love to see this for other common cell complexes. How can one generate an in vivo microtubule with 13 protofilaments and a proper seam from 1TUB for example? What rotation per y translation might a user need to enter to generate an actin filament from an actin monomer? A handful of filamentous files exist in the PDB, but animators can benefit from viewport and render time efficiencies afforded by modern software by cloning a single molecule rather than rendering coordinates from the thousands of copies of 1TUB needed to generate a lengthy microtubule. A. Gaël McGill: Basic collision detection is also not easy to implement at the moment (whether between different parts of the same continuous mesh or between meshes). Some way of integrating electrostatic forces would also be amazing! Better simulation tools would also help us create molecular vistas with some semblance to what is happening in vivo. By simulation I don't mean at the same atom-by-atom level that molecular dynamics offers, but something that would drive the stochastic behavior of numerous molecules within a defined volume or environment, for example. Finally, an area that is ripe for exploration: we need to tap into the full promise of educational gaming and interactive environments by harnessing the power of modern gaming engines. In many cases, the digital assets (models, textures, rigs) used to develop high-end games are created in packages like Maya. So one could easily imagine a scenario where a lot of the work being done to create 'narrative-driven' molecular movies in Maya could be repurposed and adapted to generate interactive molecular environments for educational purposes. These questions do not appear in the printed edition of the newsletter
Q. What packages do you typically use for your molecular animation projects? 1Representation of viruses in the remediated PDB archive (2008) Acta Cryst. D64: 874-882 doi: 10.1107/S0907444908017393 |


E-mail: info@rcsb.org • Web: www.pdb.org • FTP: ftp.wwpdb.org
The RCSB PDB is a member of the wwPDB (www.wwpdb.org)